|
|||||||||||
|
IV. Ecosmomics: Independent Complex Network Systems, Computational Programs, Genetic Ecode Scripts3. Whole Genome Regulatory Systems: DNA + AND = ANN/DAN
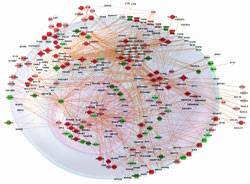
As our emergent sapiensphere proceeds to learn on her/his own, we add a 2015 section akin to other natural Systems units because genetic phenomena, with the internal components now identified and sequenced, are being seen and treated by the same informational, self-organized, complex modular network criticalities as everywhere else. In this Omics era whole genomes appear as a prime exemplar of this independent mathematical source. Nodal DNA nucleotides + connective AND networks array across dynamic scales to inform and guide lifes evolutionary gestation, each myriad creature, ourselves, and a celestial ovoGenesis. Gene regulatory networks (GRNs) which connect nucleotides and biomolecules are receiving concerted notice, as references attest. Aguirre, Jacobo, et al. On the Networked Architecture of Genotype Spaces and Its Critical Effects on Molecular Evolution. arXiv:1804.06835. In April 2018, Charles III University of Madrid, Interdisciplinary Group of Complex Systems theorists Aguirre, Pablo Catalan, Jose Cuesta, and Susanna Manrubia post a 48 page, 205 reference inclusive synthesis that ranges across statistical physics, multiplex networks, nonlinear complexities, which further takes on a computational guise. In this contribution, a whole dynamic genome serves as an exemplar, but as these integrations proceed they are found to apply in kind from quantum and cerebral realms to ecosystems and cultural societies (as this whole site seeks to document). Evolutionary dynamics is often viewed as a subtle process of change accumulation that causes a divergence among organisms and their genomes. However, this interpretation is an inheritance of a gradualistic view that has been challenged at the macroevolutionary, ecological, and molecular level. Actually, when the complex architecture of genotype spaces is taken into account, the evolutionary dynamics of molecular populations becomes intrinsically non-uniform, sharing deep qualitative and quantitative similarities with slowly driven physical systems: non-linear responses analogous to critical transitions, sudden state changes, or hysteresis, among others. Furthermore, the phenotypic plasticity inherent to genotypes transforms classical fitness landscapes into multiscapes where adaptation in response to an environmental change may be very fast. The quantitative nature of adaptive molecular processes is deeply dependent on a networks-of-networks multilayered structure of the map from genotype to function that we begin to unveil. (Abstract) Albors, Carlos, et al. A Phylogenetic Approach to Genomic Language Modeling.. arXiv:2503.03773. We cite this entry by UC, Berkeley compututational biologists and statisticians including Yun Song and Gonzalo Benegas as an example of active endeavors to work out a viable, reciprocal integration of Genome association studies and Large language methods. As AI capabilities continue to expand, this novel linguistic aspect is seen bring enhanced insights and benefits. See also A DNA language model based on multispecies alignment predicts the effects of genome-wide variants by Gonzalo Benegas, et al in Nature Biotechnology. (January 2025) for more from this group.
Angelin-Bonnet, Olivia, et al. Gene Regulatory Networks: A Primer in Biological Processes and Statistical Modelling. arXiv:1805.01098. Massey University, New Zealand mathematical geneticists present a tutorial chapter for a forthcoming Springer volume with the first title. As the Abstract notes, in contrast to the 20th century central dogma of only genes, into the 2010s, an equal presence of these pervasive linkages play a prime role in genomic contributions. Modelling gene regulatory networks not only requires a thorough understanding of the biological system depicted but also the ability to accurately represent this system from a mathematical perspective. Throughout this chapter, we aim to familiarise the reader with the biological processes and molecular factors at play in the process of gene expression regulation. We first describe the different interactions controlling each step of the expression process, from transcription to mRNA and protein decay. In the second section, we provide statistical tools to accurately represent this biological complexity in the form of mathematical models. Amongst other considerations, we discuss the topological properties of biological networks, the application of deterministic and stochastic frameworks and the quantitative modelling of regulation. (Abstract) Baysoy, Alex, et al. The technological landscape and applications of single-cell multi-omics. Nature Reviews Molecular Cell Biology,. September, 2023. We cite this work by Yale University biomedical engineers among several later 2023 entries which describe advanced Earthuman abilities to probe ever deeper into lifes implicate genomic source code prescriptive occasion. See also Human-specific genetics by Alex Pollen, et al in Nature Reviews Genetics (September 2023) and earlier Statistical mechanics meets single-cell biology by A. Teschendorff and A. Feinberg in NRG (April 2021). Single-cell multi-omics methods distinguish cell states and activities by integrating single-modality omics that profile the transcriptome, genome, epigenome, epitranscriptome, proteome, metabolome and other omics modes. Here we discuss how multi-omics has been adapted and improved over the past decade by an optimization framework of throughput, resolution, modality integration, uniqueness and accuracy. We highlight their benefit in cell lineage tracing, tissue-specific and cell-specific atlas production, tumour immunology and cancer genetics. (Excerpt) Bouyloukos, Costas, et al. GREAT: A Web Portal for Genome Regulatory Architecture Tools. Nucleic Acid Research. 44/Web Server Issue, 2016. We choose this sample entry by CNRS, University of Paris, geneticists including Francois Kepes from this 2016 update of worldwide endeavors to develop software to treat, sequence, and catalog whole genomes. A companion 2016 Database issue, each accessible in full, contains more reports from the USA, Europe, and Asia. Please note the second quote for its observation of an innately ordered genomic organization. GREAT (Genome REgulatory Architecture Tools) is a novel web portal for tools designed to generate user-friendly and biologically useful analysis of genome architecture and regulation. The online tools of GREAT are freely accessible and compatible with essentially any operating system which runs a modern browser. GREAT is based on the analysis of a genome layout - defined as the respective positioning of co-functional genes - and its relation with chromosome architecture and gene expression. (Abstract excerpt) Capozziello, Salvatore, et al. The Chern-Simons Current in Systems of DNA-RNA Transcriptions. Annalen der Physik. 530/4, 2018. In this European physics journal since 1799 (search Physik), a global research group from the University of Naples, Slovak Academy of Sciences, Chulalongkorn University, Bangkok, and National Technical University of Athens scope out this recognition of mathematical principles being found in active effect for living phenomena. An especial case is their formative presence in genomic activities. Some 319 years later, such worldwide collaborations seem at last to be quantifying an integral synthesis of emergent life and persons with a truly conducive ecosmos uniVerse. A Chern‐Simons current, coming from ghost and anti‐ghost fields of supersymmetry theory, can be used to define a spectrum of gene expression in new time series data where a spinor field, as alternative representation of a gene, is adopted instead of using the standard alphabet sequence of bases A, T, C, G, U. We give a general discussion on the use of supersymmetry in biological systems, discuss the codon and anti‐codon ghost fields and develop an algebraic construction for the DNA area which does not seem active in biological systems. Finally, we plot a time series of genetic variations of viral glycoprotein gene and host T‐cell receptor gene by using a gene tensor correlation network related to the Chern‐Simons current. An empirical analysis of genetic shift, in host cell receptor genes with separated cluster of gene and genetic drift in viral gene, is obtained by using a tensor correlation plot over time series data derived as the empirical mode decomposition of Chern‐Simons current. (Abstract edits) Cattani, Carlo and Gaetano Pierro. On the Fractal Geometry of DNA by the Binary Image Analysis. Bulletin of Mathematical Biology. Online June, 2013. We cite this technical paper by University of Salerno geneticists as an example of the extent that genomes are now found to be equally defined by such dynamic, self-similar regulatory networks. One might well imagine DNA/AND. Moreover these are the same archetypal topologies that distinguish every other strata and instance in nature. In some recent papers, the fractal nature of nucleotide distribution in DNA has been investigated in order to classify/compare DNA sequences and to single out some singularities in the nucleotide distribution. Almost all these papers are motivated by the hypothesis that although the nucleotide distribution looks like a random distribution, some parameters and typical patterns suggest to us that there might exist some fractal rules behind, which enable the chaotic organization of nucleotides. Furthermore, this structural organization seems to obey recursive laws, which are fractal-like. At the moment, we can assume that the following two axioms hold. (1) Nucleotides are distributed according to some hidden rules. (2) These rules are based on some recursive fractal law so that the nucleotide distribution obeys some fractal-like organization. (2) Chapman, Robert, et al. EcoGenomics: An Analysis of Complex Systems via Fractal Geometry. Integrative and Comparative Biology. 46/6, 2006. A salient research contribution to the total rethinking of genetic informational systems. In this regard, microarray transcription profiles are more than a collection of DNA strands, rather they are characterized by a dynamic regularity which can be explained by artificial neural networks and fractal analysis. Just as scalar ecological patterns such as species abundance are discernible through nonlinear methods, so do genomes express the same nested forms and processes. In perspective, a new nature is being untangled as no longer composed of postage stamp fragments but graced by a universal creative geometry. And this achievement need be recognized as an awesome discovery by cerebral humankind of a natural genesis. Ecogenomics is a convenient descriptor for the application of advanced molecular technologies to studies of organismal responses to environmental challenges in their natural settings. (902) Fractals are also self-similar, meaning that they contain copies of themselves within themselves, and, while they may not exhibit the same details at all scales, they exhibit the same type of structure. Hence, they are considered to be scale invariant. (903) Chia, Nicholas and Nigel Goldenfeld. Statistical Mechanics of Horizontal Gene Transfer in Evolutionary Ecology. Journal of Statistical Physics. 142/1287, 2011. As per the quotes, University of Illinois, Institute for Genomic Biology, physicists scope out commonalities between disparate biological phenomena, lately seen as many element, interactive complex systems, with seemingly inorganic many-body, condensed matter. The article concludes that very generic processes appear to repeat at every scalar instance, which would be much indicative of a genetic essence at work. The biological world, especially its majority microbial component, is strongly interacting and may be dominated by collective effects. In this review, we provide a brief introduction for statistical physicists of the way in which living cells communicate genetically through transferred genes, as well as the ways in which they can reorganize their genomes in response to environmental pressure. We discuss how genome evolution can be thought of as related to the physical phenomenon of annealing, and describe the sense in which genomes can be said to exhibit an analogue of information entropy. As a direct application of these ideas, we analyze the variation with ocean depth of transposons in marine microbial genomes, predicting trends that are consistent with recent observations using metagenomic surveys. (Abstract, 1287) Cortini, Ruggero, et al. The Physics of Epigenetics. arXiv:1509.04145. In a paper to appear in the Review of Modern Physics, French scientists at Sorbonne and CNRS laboratories, including Annick Lesne and Jean-Marc Victor, contribute to the vital 21st century synthesis of life with its contextual milieu by showing how physical phenomena such as complex systems are a basis for extra-genomic transcriptions. A working title definition is given in the second quote. Whatever to then make of such a fecund cosmic ground, which seems to be graced by a primal genetic essence? In higher organisms, all cells share the same genome, but every cell expresses only a limited and specific set of genes that defines the cell type. During cell division, not only the genome, but also the cell type is inherited by the daughter cells. This intriguing phenomenon is achieved by a variety of processes that have been collectively termed epigenetics: the stable and inheritable changes in gene expression patterns. This article reviews the extremely rich and exquisitely multi-scale physical mechanisms that govern the biological processes behind the initiation, spreading and inheritance of epigenetic states. Strikingly, to achieve stability and heritability of epigenetic states, cells take advantage of many different physical principles, such as the universal behavior of polymers and copolymers, the general features of non-equilibrium dynamical systems, and the electrostatic and mechanical properties related to chemical modifications of DNA and histones. By putting the complex biological literature under this new light, the emerging picture is that a limited set of general physical rules play a key role in initiating, shaping and transmitting this crucial "epigenetic landscape". (Abstract excerpts) Cowen, Lenore, et al. Network Propagation: A Universal Amplifier of Genetic Associations. Nature Reviews Genetics. 18/9, 2017. We note this paper by geneticists Cowen, Tufts University, with Trey Ideker, UC San Diego, Ben Raphael, Princeton, and Roded Sharan, Tel Aviv University, for its emphasis on the active presence in genomic systems of network multiplexities. In regard, as everywhere else, natures independent, tendency to form node and link structures are similarly manifest in complex genomes. Biological networks are powerful resources for the discovery of genes and genetic modules that drive disease. Fundamental to network analysis is the concept that genes underlying the same phenotype tend to interact; this principle can be used to combine and to amplify signals from individual genes. Recently, numerous bioinformatic techniques have been proposed for genetic analysis using networks, based on random walks, information diffusion and electrical resistance. These approaches have been applied successfully to identify disease genes, genetic modules and drug targets. In fact, all these approaches are variations of a unifying mathematical machinery network propagation suggesting that it is a powerful data transformation method of broad utility in genetic research. (Abstract) Daban, Joan-Ramon. Soft-Matter Properties of Multilayer Chromosomes. Physical Biology. 18/5, 2021. An Autonomous University of Barcelona biochemist offers a latest orientation to the apparently autonomous properties of natures fertile materiality, by which to apply them to active genomic phenomena. In each case, self-organizing processes serve to generate vital forms: Chromatin structure is locally dynamic like a liquid crystal fluid. Altogether another example of mid 2021 revolutionary discoveries. This perspective will identify relationships between the structural and dynamic properties of chromosomes as states of soft-matter systems. Our previous studies, using transmission electron microscopy, atomic force microscopy, and cryo-electron tomography, suggested that metaphase chromosomes have a multilayered structure. The self-assembly of multilayer chromatin plates suggests that metaphase chromosomes are self-organized hydrogels with an internal liquid-crystal order produced by the stacking of chromatin layers along the chromosome axis. The spontaneous assembly of large structures has been studied in different soft-matter systems and the self-organization of chromosomes could be justified by the interplay between weak interactions of repetitive nucleosome building blocks and thermal fluctuations. Furthermore, at the end of mitosis, condensed chromosomes undergo a phase transition into a more fluid structure. (Abstract excerpt)
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
HOME |
TABLE OF CONTENTS |
Introduction |
GENESIS VISION |
LEARNING PLANET |
ORGANIC UNIVERSE |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||